
-
CENTRES
Progammes & Centres
Location
 PDF Download
PDF Download 
Chitra Pattabiraman, Farhat Habib and Krishnapriya Tamma, “Perspectives on SARS-CoV-2 Strains,” ORF Issue Brief No. 365, May 2020, Observer Research Foundation.
Introduction
The ongoing pandemic of the coronavirus disease 2019 (COVID-19) is caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2).[1] It is an RNA (ribonucleic acid) virus, with a genome of length 29,903 base pairs (See Box 1). The genome is organised into 11 genes that code for different proteins (See Fig. 1), such as the Spike protein.[2]
Researchers across the world have sequenced the genome of the virus using samples from infected individuals. As of early May 2020, over 17,000 genomes were available in public databases (GISAID, NCBI).[3],[4] This timely sharing of data has allowed researchers to compare these sequences,[5] revealing differences between the sequences that arose due to mutations (See Box 1). Most mutations do not result in changes to the virus; some can be deleterious and others result in positive changes. Mutations have to be taken into consideration while designing ways to detect a virus, developing vaccines, or testing potential drugs—this is because some mutations can lead to false positives in diagnostic tests, lack of response to vaccine, and development of resistance to a drug. Mutations that alter the property of the virus can give rise to biologically distinct variants called “strains” (See Box 5). At the time of writing this brief, there is no evidence that the SARS-CoV-2 sequences have any biological differences, indicating that only one strain of this virus is currently in circulation. [6]
Figure 1: Organisation of the SARS-CoV-2 Genome

Note: Genes are shown in yellow, the resulting peptides are shown in green, untranslated regions are shown in red and purple. An early sequence from Wuhan, China (NC_045512) has been used to make this schematic using Geneious (Ver. 2020.0).
The S and L “Types” of SARS-CoV-2
In February 2020, Tang et al. published an analysis of 103 SARS-CoV-2 genomes.[7]The genomes were clustered into two groups, based on two mutations. One of these was an amino acid change (from Serine to Leucine) at position 84 (See Box 3) in the ORF8 region, causing a slightly different protein to be produced. The two clusters of genomes were labelled S (Serine) and L (Leucine) types. The L type was reported to be more prevalent amongst the viruses being studied. Based on this, the researchers termed it an “aggressive type.” However, this classification was questioned by the scientific community,[8],[9] and Tang et al. conceded that without sufficient clinical and/or laboratory studies to confirm the hypothesis, “higher frequency” was a more appropriate term than “aggressive.” Unfortunately, the damage had already been done as the original article had been picked up by the international media.
A “Mutant” SARS-CoV-2
Towards the end of April 2020, Korber et al. analysed 4,535 publicly available (GISAID) SARS-CoV-2 genomes and presented their findings in the pre-print server, bioRxiv.[10] They focused on the part of the genome that codes for the Spike protein (See Fig. 1), which interacts with its receptor on human cells, allowing the virus to enter the cell (See Box 4).[11] The researchers found that at position 614 of the protein, there was a change from Aspartic Acid to Glycine (D614G), compared to the reference strain, resulting in a variant Spike protein. Further, the frequency of this mutation was increasing across the world. Since the mutation is on the protein that aids virus entry into the host cell, the researchers posited that this change might influence the ability of the protein to bind to the ACE2 receptor and that the mutation might impact the way the virus is seen and recognised by the immune system. While these are valid hypotheses, they currently remain untested. Finally, their analyses indicate that when viruses containing this mutation enter a population, they become the dominant virus in that region.
The Current Consensus on SARS-CoV-2 Strains
A “strain” for a virus (See Box 5) must have, at the very least, distinct biological properties, e.g. different growth rates and stability in different environments. Thus, virus isolates of SARS-CoV-2 do not qualify, since they have no documented biological differences.[12]
However, the analysis of global sequencing data suggested that based on sequence difference, SARS-CoV-2 genomes can be classified into at least 10 distinct groups. A phylogenetic tree depicts the relationships between the different sequences (See Fig. 2). A clade is a group of highly related sequences that share a common ancestor, i.e. they are part of one branch. In the SARS-CoV-2 phylogeny, many of the original isolates from China are part of the B clade. Isolates from across the world belong to the A clade (using the same nomenclature as used by nextstrain.org). The phylogeny is updated as new sequences arrive, and the most recent analysis (early May 2020) of the data (5,234 complete genomes) by 386 groups across the world shows that the A2a is the most commonly sequenced clade (See Fig 2). One of the mutations defining the A2a clade is the same as the one described by Korber et al., i.e. D614G on the Spike protein.
Majumder et al. have come to a similar conclusion about the global prevalence of A2a (unpublished results).[13] However, Dr. Majumder stated in an interview that the available data from Asia does not show a clear dominance of the A2a clade.[14]
Figure 2: Phylogeny of 5,234 Complete Genomes
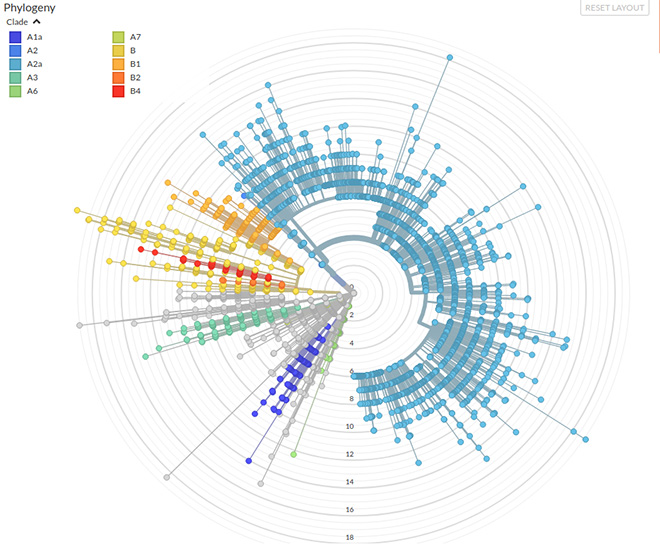
A2a Clade Transmission
Four primary factors limit the ability of researchers to ascertain whether the A2a clade is transmitting better than other clades. These must be considered while analysing transmission rates of different clades of SARS-CoV-2 in the global setting.
The Implication of a Mutating Virus
Mutations can affect the efficacy of sequence-based diagnostic methods and impose restrictions on vaccine design.[20] The most commonly used diagnostic tool targets specific regions of the genome, and if a mutation occurs in this region, the test may lead to a false negative result. Furthermore, sequence-based vaccines may have to incorporate the D164G mutation and account for its effects. Sequencing efforts all over the world are flagging regions that are mutating to guide diagnostics and vaccine design.
Mutations are a part of the natural life cycle of any virus, particularly RNA viruses. The many mutations observed in the SARS-CoV-2 sequences are therefore expected.[21] However, it is unclear whether these mutations are meaningful, i.e. if they change the biological properties of the virus. Studies to evaluate this are yet to be conducted. While it is plausible that some mutations may allow the virus to spread more effectively, it is premature to conclude that such mutations are driving the current global spread of the virus.
Conclusion
Analyses of genome sequences from across the globe have revealed the presence of different SARS-CoV-2 clades. However, there is no experimental evidence to suggest a difference in aggressiveness amongst these. Moreover, the effects of the observed mutations in SARS-CoV-2 on the properties of the virus are yet to be evaluated in clinical or experimental studies. Since multiple factors influence viral spread, the mere prevalence of a virus clade cannot be used as a proxy for biological traits such as increased transmission and disease severity. Some countries are affected more than others due to a complex mix of the biology of the virus and the behaviour of the infected and susceptible population. An exhaustive list of the major factors can only emerge over time, as more data becomes available.
Disclaimer: Some of the research work cited here has not yet been vetted by other scientists in a formal manner (peer-review). The information presented in this article is current as of 18 May 2020.
Box 1: The Genome of SARS-CoV-2SARS-CoV-2 is an RNA virus, and its genome is made up of a unique arrangement of four ribonucleotides—Adenine (A), Uracil (U), Guanine (G), Cytosine (C)—approximately 30,000 bases long. The genome can be thought of as a lengthy paragraph. When a virus infects a cell, it makes copies of itself. This requires copying out the paragraph, wherein mistakes (i.e. mutations) can happen. The more the number of copies, the higher the chances of accumulating ‘mutations’. Some of these can change the meaning, others may render the paragraph meaningless, whereas many errors (such as typos) may not affect the meaning at all. |
Box 2: Do viruses change in a way that they transmit better or cause more severe disease?Avian Influenza Virus – Studies have shown that a small number of mutations in the avian influenza viruses can make them transmit amongst mammals.[22]Additionally, a small number of mutations have been predicted to increase the chances of spillover into human populations and transmission amongst humans.[23] It is posited that something similar occurred in an ancestor of SARS-CoV-2, allowing it to jump into humans in 2019. SARS – When Severe Acute Respiratory Syndrome (SARS) virus emerged in 2003, a 29-nucleotide deletion was observed. It was initially postulated to have a positive effect; however, later studies demonstrated a deleterious effect of this mutation on the virus.[24] Zika Virus – There are two lineages (African and Asian) of the mosquito-borne Zika virus, based on the differences in their genetic sequences. However, the African lineages are not known to cause birth defects, whereas the Asian lineages have caused outbreaks of microcephaly in many parts of the world. There is evidence to suggest that the Asian lineage acquired a single mutation, making it more pathogenic than the older Zika viruses of the same lineage.[25]This strain was responsible for the devastating outbreak in Brazil in 2015. Ebola Virus -A mutation in the Ebola virus was reported in 2015 and thought to have given rise to a more aggressive strain.[26] However, while initial studies provided strong evidence for these differences, these were not replicated in animal models. Thus, it has proven difficult to interpret the effect of this mutation.[27] |
Box 3: The Building Blocks of the BodyProteins are the key functional molecules in biology: most enzymes and many hormones and toxins are proteins. They are chains of smaller units called amino acids. There are 20 amino acids, and each has specific chemical properties—what it can bind, whether they can dissolve in water, etc. Thus, the structure and function of a protein is determined by the specific sequence and identity of the amino acids that constitute it. Amino acids are coded by the nucleotide sequences of the genetic material. Non-synonymous mutations lead to a change in the amino acid, while synonymous mutations do not. Changes in the amino acids in the protein sequence may change the structure and function of the protein. |
Box 4: The Spike ProteinThe Spike (S) protein sits on the surface of the virus and gives it a crown-like appearance (hence the name coronavirus). The protein can pair with the ACE2 (Angiotensin Converting Enzyme 2) protein on human cells.[28] The binding of these two proteins from two different organisms opens a pathway for the virus to enter the human cell. Therefore, the S protein is important and changes to this protein might change the ease with which the virus enters a human (host) cell. Evidence from sequence comparisons suggests that changes in the S protein allowed this virus to jump species and successfully infect humans.[29] |
Box 5: What is a “strain” of a virus?A “strain” can be defined as a variant of a virus species that is distinctly recognisable and possesses some unique phenotypic characteristics, which are stable under natural conditions, e.g. antigenic properties, disease manifestation or host range.[30] The condition of “stable genetic difference” requires that the strain differences are stable across generations, possibly due to natural selection. According to this definition, if two viruses show only genotypic differences without phenotypic differences, they will not qualify as different strains. On the other hand, if they show phenotypic differences even with few mutations, they will qualify as different strains. Thus, genomic information alone, while very important, is insufficient in determining virus classification, reconstructing evolution, or even understanding pathogenicity. |
Box 6: How fast is this virus changing?Current estimates for the mutation rate of SARS-CoV-2 are about one mutation in two weeks.[31] This is calculated from the estimated evolutionary rate of 8×10-4 substitutions/site/year.[32] This rate is close to those of SARS and MERS, two viruses of the same family.[33] |
Box 7: Phylogenetic TreeA phylogenetic tree is a diagrammatic representation of the relationship between different taxa. Typically, genetic data (sequences or genomic data) is used to reconstruct phylogenetic trees. The overall shape of the tree is called its “topology,” and the tips correspond to the “taxa” whose relationship is being examined. These often correspond to different species, but can also correspond to virus strains, individuals of a species, genera, or other higher levels of organisation. The tips are connected to each other by branches. Two adjacent tips are connected by a “node,” which corresponds to the recent shared common ancestor. The taxa (or tips) that share a common ancestor are considered “sisters.” If all taxa share a common ancestor, and all descendants are included in the group, it is called a “monophyletic group.” An important component of a phylogenetic tree is a “clade,” which is a monophyletic group of taxa that includes the most recent common ancestor of all the members (i.e. all the descendants). A clade can be defined at any level in a phylogenetic tree. |
(The authors thank Prof. Nagasuma Chandra for initial discussions and reading material suggestions for this brief. We would also like to acknowledge researchers from across the world who have deposited sequences in GISAID and the nextstrain.org team for sharing their analysis.)
About the Authors
The authors are members of a multidisciplinary COVID-19 study circle based in India. Chitra Pattabiraman is Early Career Fellow, India Alliance (DBT-Wellcome Trust), at the Department of Neurovirology, NIMHANS, Bangalore; Farhat Habib is Director of Data Science at TruFactor (an InMobi Group Company), Bangalore; and Krishnapriya Tamma is Assistant Professor, School of Arts and Sciences, Azim Premji University, Bangalore.
Endnotes
[1] Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. “A novel coronavirus from patients with pneumonia in China, 2019,” N Engl J Med (2020). https://doi.org/10.1056/NEJMoa2001017.
[2] Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. “Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding,” Lancet (2020). https://doi.org/10.1016/S0140-6736(20)30251-8.
[3] GISAID. URL: https://www.gisaid.org/
[4] NCBI. URL: https://www.ncbi.nlm.nih.gov/
[5] nextstrain.org. URL: https://nextstrain.org/ncov
[6] “There is one, and only one strain of SARS-CoV-2”, virology.ws. (2020) URL: https://www.virology.ws/2020/05/07/there-is-one-and-only-one-strain-of-sars-cov-2/
[7] Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. “On the origin and continuing evolution of SARS-CoV-2,” Natl Sci Rev (2020). https://doi.org/ 10.1093/nsr/nwaa036.
[8] MacLean OA, Orton RJ, Singer JB, Robertson DL. “Response to “On the origin and continuing evolution of SARS-CoV-2” (2020). URL: http://virological.org/t/response-to-on-the-origin-and-continuing-evolution-of-sars-cov-2/418/11.
[9] MacLean OA, Orton RJ, Singer JB, Robertson DL. “No evidence for distinct types in the evolution of SARS-CoV-2,” Virus Evol (2020);6:. https://doi.org/10.1093/ve/veaa034
[10] Korber B, Fischer W, Gnanakaran SG, Yoon H, Theiler J, Abfalterer W, et al.“Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2,” BioRxiv (2020).
[11] Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. “SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor,” Cell (2020).
[12] “There is one, and only one strain of SARS-CoV-2”, virology.ws. (2020).
[13] “Listen: The Science and Mysteries of How the New Coronavirus Is Evolving”, 9 May 2020.
[14] “Ep. 136: The SARS-COV-2 Virus: Mutations & Evolution”, 8 May 2020.
[16] Bruinen de Bruin Y, Lequarre A-S, McCourt J, Clevestig P, Pigazzani F, Zare Jeddi M, et al. “Initial impacts of global risk mitigation measures taken during the combatting of the COVID-19 pandemic“, Saf Sci (2020);128:104773.
[17] CDC Forecasts (2020).
[18] Ferguson N, Laydon D, Nedjati Gilani G, Imai N, Ainslie K, Baguelin M, et al.Impact of non-pharmaceutical interventions (NPIs) to reduce COVID19 mortality and healthcare demand. n.d.
[19] Lv M, Luo X, Estill J, Liu Y, Ren M, Wang J, et al. “Coronavirus disease (COVID-19): a scoping review,” Eurosurveillance (2020);25:2000125.
[20] Phelan J, Deelder W, Ward D, Campino S, Hibberd ML, Clark TG. “Controlling the SARS-CoV-2 outbreak, insights from large scale whole genome sequences generated across the world“, BioRxiv (2020):2020.04.28.066977.
[21] Sidney M. Bell, Emma Hodcroft, Nicola Müller, Cassia Wagner, James Hadfield, Richard Neher TB. “Genomic analysis of COVID-19. Situation report 2020-05-15”(2020)
[22] Herfst S, Schrauwen EJA, Linster M, Chutinimitkul S, Wit E de, Munster VJ, et al. “Airborne Transmission of Influenza A/H5N1 Virus Between Ferrets“, Science (2012);336:1534–41.
[23] “Reconstruction of 1918-like avian influenza virus stirs concern over gain of function experiments”,(2014).
[24] Muth D, Corman VM, Roth H, Binger T, Dijkman R, Gottula LT, et al.“Attenuation of replication by a 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission“, Sci Rep(2018);8:15177.
[25] Yuan L, Huang X-Y, Liu Z-Y, Zhang F, Zhu X-L, Yu J-Y, et al. “A single mutation in the prM protein of Zika virus contributes to fetal microcephaly“, Science (2017);358:933–6.
[26] Diehl WE, Lin AE, Grubaugh ND, Carvalho LM, Kim K, Kyawe PP, et al. “Ebola Virus Glycoprotein with Increased Infectivity Dominated the 2013–2016 Epidemic,” Cell (2016);167:1088-1098.e6.
[27] Marzi A, Chadinah S, Haddock E, Feldmann F, Arndt N, Martellaro C, et al.“Recently Identified Mutations in the Ebola Virus-Makona Genome Do Not Alter Pathogenicity in Animal Models“, Cell Rep (2018);23:1806–16.
[28] Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. “SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor“, Cell (2020).
[29] Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. “The proximal origin of SARS-CoV-2“, Nat Med (2020).
[30] Kuhn JH, Bao Y, Bavari S, Becker S, Bradfute S, Brister JR, et al. “Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family Filoviridae“, Arch Virol (2013);158:301.
[31] Louis du Plessis. Temporal signal of nCoV-2019 based on 30 genomes, (2020).
[32] Rambaut A. Phylodynamic Analysis | 176 genomes | 6 Mar 2020’, (2020).
[33] MacLean OA, Orton RJ, Singer JB, Robertson DL. “No evidence for distinct types in the evolution of SARS-CoV-2“, Virus Evol (2020);6:.
The views expressed above belong to the author(s). ORF research and analyses now available on Telegram! Click here to access our curated content — blogs, longforms and interviews.

Dr. Chitra Pattabiraman is a virologist/molecular biologist who uses genomic tools (sequencing) to identify and characterize pathogens. She obtained her Integrated MSc- PhD in Life ...
Read More +
Farhat Habib is Director of Data Science at TruFactor (an InMobi Group Company) Bangalore
Read More +
Krishnapriya Tamma is Assistant Professor School of Arts and Sciences Azim Premji University Bangalore.
Read More +



